Pterigium colli
Pterygium colli (Cuello alado), es un pliegue epidérmico del borde externo del cuello, a manera de aleta, que va desde la implantación de las orejas hasta los hombros. Aparece típicamente en el síndrome de Noonan, pero también en otros trastornos genéticos como el síndrome de Turner, síndrome de Edwards y con menos frecuencia, el síndrome de Down.
Es un trastorno genético que produce desarrollo anormal de múltiples partes del cuerpo. Se caracteriza por una serie de síntomas y características físicas que pueden variar ampliamente en rango y severidad según los casos.
| Pterigium colli | ||
|---|---|---|
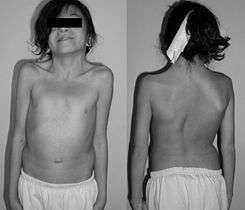 Pterigium colli en una niña de 12 años con el síndrome de Noonan. | ||
| Clasificación y recursos externos | ||
| Especialidad | Genética médica | |
| CIE-10 | Q18.3 | |
| CIE-9 | 744.5 | |
En la mayoría de los casos se transmite como un rasgo genético autosómico dominante, pero puede haber casos esporádicos. En particular, se presentan membranas en el cuello y diferentes formas del tórax que recuerdan al síndrome de Turner, por lo que siempre hay que saber diferenciarlo de este síndrome.
El síndrome de Noonan fue descrito en 1963 por Noonan y Ehmke en pacientes con estenosis valvular pulmonar, asociado a baja estatura, hipertelorismo y retardo mental moderado, entre otras alteraciones. La virtud de Jacqueline Noonan fue que además de indicar los signos clínicos mayores, observó que la cardiopatía más frecuente era la estenosis pulmonar (17 en 19 pacientes) (Noonan 1968), lo que diferenciaba esta enfermedad del síndrome de Turner, donde la cardiopatía que se presenta con mayor frecuencia es la coartación de la aorta.
Se le puede denominar de las siguientes maneras:
- Síndrome de Ullrich Noonan,
- Síndrome de Ullrich,
- Síndrome de Pseudo Turner,
- Fenotipo Turner con Cariotipo Normal
- Síndrome de Turner del Varón
- Síndrome del Pterigium Colli.
Causas e incidencia
El síndrome de Noonan afecta al menos a 1 de cada 2.500 niños. A diferencia del síndrome de Turner, los afectados por el Noonan no tienen alteraciones del cariotipo, es decir, no tienen alteración del número ni de la organización de los cromosomas, estructuras que contienen la información genética. Diferentes estudios detectaron familias en donde el síndrome aparecía en varios miembros, bajo una transmisión vertical, con rasgos diferentes de unos a otros e incluso con generaciones saltadas (dominancia irregular), pero con un predominio de herencia por vía materna. Esto estableció un patrón de herencia autosómica dominante, lo que significa que en estos casos sería necesario la presencia de la mutación ya en los genes de uno de los progenitores, y que esta mutación sería transmitida a los hijos con un 50% de probabilidad, aunque en esta enfermedad, la probabilidad de que, una vez presente, se exprese de una forma grave sería del 14%. El hecho de que algunos niños no tengan un padre con el síndrome de Noonan refleja la posibilidad de una aparición esporádica, es decir, la presencia presumible de una nueva mutación, no presente en los genes de los padres.
Desde hace unos pocos años se ha identificado el locus donde se ubica el gen que condiciona el fenotipo de al menos un gran porcentaje de personas con S. de Noonan y que se sitúa en 12q24 (Jamieson). En este emplazamiento del mapa genético se encuentra el primer gen específico identificado como posible responsable del síndrome de Noonan, denominado PTPN11.
Este único gen no puede explicar todos los casos, por lo que se espera el descubrimiento de otros genes que producen este síndrome.
Síntomas
Las anomalías que se ven con más frecuencia incluyen formación de membranas en el cuello, cambios en el esternón (generalmente un tórax hundido llamado tórax excavado), anomalías faciales y enfermedad cardíaca congénita (especialmente estenosis pulmonar). En lenguaje coloquial, las anomalías más características del síndrome de Noonan son:
- la estatura corta
- cuello ancho
- cardiopatía (enfermedad del corazón), siendo la estenosis pulmonar la más frecuente.
- hipertelorismo (aumento de la separación de los ojos)
- malformaciones del esternón con pectus carinatum (tórax en quilla) y excavatum (tórax en embudo)
- tórax ancho
- mamas separadas y bajas
- pterigium colli (pliegue del borde externo del cuello que va desde la implantación de las orejas hasta los hombros) y pterigium axilar (membrana cutánea que la axila)
- aspecto típico de la cara con filtrum (surco vertical en el centro del labio superior) marcado
- fisuras palpebrales antimongoloides
- paladar ojival (paladar en forma de bóveda)
- micrognatia (mandíbula anormalmente pequeña)
- orejas displásicas (displasia es el desarrollo anómalo de tejidos u órganos), de implantación baja y rotadas
- pliegues en la piel de la nuca
- implantación baja del cabello en la zona posterior
- párpados gruesos
- epicantus (dobleces adicionales de la piel en las esquinas internas de los ojos)
- exoftalmos (protrusión anormal del globo del ojo) y ptosis palpebral (párpados caídos)
- Suelen presentar malformaciones del corazón e hipoplasia (desarrollo incompleto o defectuoso) o aplasia (ausencia de desarrollo) de vasos sanguíneos y linfáticos, alteraciones de las plaquetas y de los factores de la coagulación de la sangre e hipertermia maligna.
Se presenta retardo mental leve en aproximadamente el 25% de los casos y pérdida auditiva variable. Generalmente se presenta un retardo en la pubertad y los hombres pueden tener testículos no descendidos y un pene pequeño.
Signos y exámenes
El diagnóstico del síndrome de Noonan es fundamentalmente clínico: se diagnostican como tales a todos aquello que cumplan las características fenotípicas que definen este síndrome. Un sencillo examen físico, por ejemplo, puede mostrar un pliegue extra de la piel por encima de los ojos (pliegues del epicanto) que también pueden parecer inclinados (inclinación palpebral antimongoloide), o cualquiera de las demás alteraciones faciales o corporales de este síndrome. Los brazos pueden estar sostenidos en un ángulo inusual (cúbito valgo).
Es importante descartar la existencia de signos de enfermedad cardíaca congénita (a menudo estenosis pulmonar y ocasionalmente defecto del tabique auricular), para lo se podrán realizar diferentes pruebas como un electrocardiograma, o pruebas de imagen como una radiografía de tórax o un ecocardiograma.
Para detectar las alteraciones en la hemostasia (control del sangrado) que pueden presentarse, basta con una sencilla analítica que revela el bajo recuento de plaquetas o alteraciones en los exámenes de coagulación y en la medición de los niveles de factores específicos de coagulación en la sangre (factores XI-XIII).
La evaluación genética puede identificar mutaciones causales en el gen PTPN1 y se utiliza un cariotipo para confirmar que una niña no tiene un trastorno de apariencia similar, el síndrome de Turner.
Diagnóstico diferencial y variantes del Síndrome de Noonan
Van del Burgt clasifica 6 rasgos clínicos en Mayores y Menores. Según esta clasificación, un individuo presenta este síndrome si reúne la cara típica, más otro signo mayor o dos menores. Igualmente si los rasgos faciales son sugestivos y a ellos se suman dos criterios mayores o tres signos menores.
RASGOS MAYORES MENORES
CARA Característica Sugerente
CORAZÓN Estenosis pulmonar y/o ecg típico Otras cardiopatías
ESTATURA Menor del percentil 3 Menor del percentil 10
TÓRAX Pectum carinatum / excavatum Ancho
HISTORIA FAMILIAR Padre o madre con s. de noonan Ídem con algún rasgo
OTROS Los tres criterios en niños (dos en niñas): déficit mental /criptorquidia/ displasia linfática Uno de ellos
El diagnóstico prenatal solamente cabe en forma de sospecha ante la presencia de higroma quístico nucal o linfedema, cuando el cariotipo es 46, XX o 46, XY, pero no existen criterios ultrasonográficos para la identificación precoz (Achiron).
En las niñas, fundamentalmente con el S. de Turner y en ambos sexos es preciso excluir sobre todo la embrio-fetopatía por alcohol y por primidona, amén de otros cuadros con algunos rasgos comunes como el S. de Aarskog o la trisomía del brazo corto del cromosoma 8.
Rasgos apreciables en la forma clásica del Noonan a los que se suman otros se dan en los síndromes de:
- S. de Watson (rasgos de NF1 y estenosis pulmonar),
- LEOPARD entiginosis, ECG con signos de anomalía de conducción, hipertelorismo Ocular, estenosis Pulmonar, Anomalías genitales, Retraso en el crecimiento, sordera)
- Cardio-Facio-Cutáneo (CFC) (frecuente retraso mental y del crecimiento, hipotricosis, eczema resistente al tratamiento)
Lo que complica la precisión del diagnóstico en situaciones como la descrita por Leichtman en que una madre con rasgos de Noonan tuvo una niña con CFC.
Tras el nacimiento de un afectado, si se trata de una forma esporádica en donde los progenitores no muestran rasgos de S. de Noonan, el riesgo empírico de recurrencia es el 5%. En los casos de herencia autonómica dominante, con un padre ya afectado, aunque sea de forma leve, la recurrencia para siguientes hijos es del 50% considerándose que para que se manifieste como forma grave es el 14% (Sharland). Si los cónyuges tuvieran una relación de consanguinidad, el riesgo de recurrencia es el 25% con la consideración de la mayor probabilidad de que el síndrome curse con miocardiopatía hipertrófica obstructiva.
Tratamiento y seguimiento
No hay un tratamiento único para el síndrome de Noonan. El tratamiento se centra en los problemas que se presentan. Tras el diagnóstico neonatal fenotípico externo se impone la evaluación cardiológica como primera medida a seguir. Se refiere que hasta un 6% fallecen cada año por arritmia. Luego hay que centrarse en la observancia evolutiva del crecimiento estatoponderal y del desarrollo psicomotor.
Por lo demás, como cualquier otro niño aunque pueden darse dificultades para la alimentación (anorexia, reflujo gastro-esofágico...). No hay que olvidarse de la mayor frecuencia de defectos de refracción, tiroiditis, posible hipertermia maligna en caso de anestesia, hipoacusia, maloclusión dental.
El uso de la hormona de crecimiento es eficaz a corto plazo aumentando la velocidad de crecimiento aunque sin que al parecer se incremente de forma ostensible la talla final: media de + 3.1cm (-1.1 a 6.5cm). No se ha constatado riesgo de cardiomiopatía hipertrófica bajo el tratamiento en el curso de 3 años de terapéutica.
En la pubertad, máxime los varones que tuvieron criptorquidia bilateral, han de ser evaluados gonadalmente tanto clínica (desarrollo de caracteres sexuales secundarios) como hormonalmente (determinación de valores de testosterona y gonadotropinas), dada la posibilidad de hipofunción. El tratamiento con administración de testosterona en parche diario o de depósito (cada 21 días) ha de ser individualizado para por un lado evitar la osteoporosis, y por otro lado que no se de una libido potencialmente peligrosa si no se controla con una inteligencia y educación suficientes.
Pronóstico y prevención
El pronóstico esperado depende de la extensión y gravedad de los síntomas existentes. Los pacientes pueden llevar una vida normal y, si se presenta retardo mental, generalmente es leve.
Las personas con antecedentes familiares de síndrome de Noonan pueden consultar con el médico antes de tener hijos. La prevención de las complicaciones, tal como enfermedad cardíaca, depende de la detección a tiempo y el cuidado continuo de un cardiólogo.
Rasgos Clínicos del Síndrome de Noonan,[1] estudio elaborado por: Joaquín Fernández Toral,[2] Julia Barreiro Daviña,[3] Mario Pestaña García[4]
- Web de la Asociación norteamericana del Síndrome de Noonan
- Profesor Titular de Pediatría de la Universidad de Oviedo y Jefe de Sección de Genética Pediátrica del Hospital Central de Asturias
- Médico Pediatra. Hospital Central de Asturias
- Servicio de Proceso de Imágenes y Tecnologías Multimedia de la Universidad de Oviedo.