

Prevalence of Amyotrophic Lateral Sclerosis — United States, 2012–2013
Surveillance Summaries / August 5, 2016 / 65(8);1–12
 ShareCompartir
ShareCompartir
Please note: An erratum has been published for this report. To view the erratum, please click here.
Paul Mehta, MD1; Wendy Kaye, PhD2; Leah Bryan, MPH3; Theodore Larson, MS1; Timothy Copeland, BA1; Jennifer Wu, MS1; Oleg Muravov, MD, PhD1; Kevin Horton, DrPH1 (View author affiliations)
View suggested citationAbstract
Problem/Condition: Amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig’s disease, is a progressive and fatal neuromuscular disease for which no cure or viable treatment has been identified. ALS, like most noncommunicable diseases, is not a nationally notifiable disease in the United States. The prevalence of ALS in the United States during 2010–2011 was estimated to be 3.9 cases per 100,000 persons in the general population. Updated prevalence estimates are needed to help monitor disease status, better understand etiology, and identify risk factors for ALS.
Period Covered: 2012–2013.
Description of System: The National ALS Registry, established in 2009, collects data on ALS patients in the United States to better describe the incidence and prevalence of ALS, examine risk factors such as environmental and occupational exposures, and characterize the demographics of those living with ALS. To identify prevalent cases of ALS, data are compiled from four national administrative databases (maintained by the Centers for Medicare and Medicaid Services, the Veterans Health Administration, and the Veterans Benefits Administration). To identify cases not included in these databases and to better understand risk-factors associated with ALS and disease progression, the Registry also includes data that are collected from patients who voluntarily enroll and complete online surveys.
Results: During 2012 and 2013, the Registry identified 14,713 and 15,908 persons, respectively, who met the surveillance case definition of ALS. The estimated ALS prevalence rate was 4.7 cases per 100,000 U.S. population for 2012 and 5.0 per 100,000 for 2013. Due to revisions to the algorithm and use of death data from the National Death Index, an updated prevalence estimate has been calculated retrospectively for October 19, 2010–December 31, 2011. This updated estimate showed a prevalence rate of 4.3 per 100,000 population and a total of 13,282 cases. Since the inception of the Registry, the pattern of characteristics (e.g., age, sex, and race/ethnicity) among persons with ALS have remained unchanged. Overall, ALS was more common among whites, males, and persons aged 60–69 years. The age groups with the lowest number of ALS cases were persons aged 18–39 years and those aged ≥80 years. Males had a higher prevalence rate of ALS than females overall and across all data sources. These findings remained consistent during October 2010–December 2013.
Interpretation: The Registry is the only available data source that can be used to estimate the national prevalence for ALS in the United States. Use of both administrative national databases and self-report from patients enables a comprehensive approach to estimate ALS prevalence. The overall increase in the prevalence rate from 4.3 per 100,000 persons (revised) during 2010–2011 to 4.7 and 5.0 per 100,000 persons, respectively, during 2012–2013 likely is not an actual increase in the number of ALS cases. Rather, this increase might be attributed to improved case ascertainment due to the refinement of the algorithm used to identify definite ALS cases, along with an increased public awareness of the Registry. Registry estimates of ALS prevalence are consistent with findings from long-established ALS registries in Europe and from smaller-scale epidemiologic studies previously conducted in the United States.
Public Health Actions: Data collected by the National ALS Registry are being used to better describe the epidemiology of ALS in the United States and to help facilitate research. The combined approach of using national administrative databases and a self-enrollment web portal to collect data is novel and potentially could be used for other non-notifiable diseases such as Parkinson’s disease or multiple sclerosis.
Increased public awareness of the Registry might lead to more ALS cases being identified from the secure web portal (https://www.cdc.gov/als), which can ascertain cases apart from the national administrative databases. For example, in 2014, the ALS Ice Bucket Challenge, a social media–centered campaign, received extensive public visibility and created increased awareness of ALS. The Agency for Toxic Substances and Disease Registry (ATSDR) works closely with ALS advocacy and support groups, researchers, health care professionals, and others to promote the National ALS Registry and to identify all cases of ALS in the United States. In addition to estimating the prevalence of ALS, the Registry is being used to collect specimens from patient enrollees through a new biorepository, connect patient enrollees with new clinical trials and epidemiologic studies, and fund studies to help learn more about the etiology of ALS. Additional information about the National ALS Registry is available at http://www.cdc.gov/als or by calling toll-free at 1-877-442-9719.
Introduction
Amyotrophic lateral sclerosis (ALS), commonly known as Lou Gehrig’s disease, is a progressive and fatal neuromuscular disease. The majority of ALS patients die within 2–5 years of receiving a diagnosis (1). There is no known definitive cause of ALS; familial ALS, a hereditary form of the disease, accounts for 5%–10% of cases (1). No cure has been identified, and the lack of proven and effective therapeutic interventions for ALS is an ongoing challenge. Riluzole (brand name Rilutek) is currently the only drug that has been approved by the Food and Drug Administration (FDA) to treat ALS; however, it only prolongs life by approximately 2–3 months on average (2). In some patients, intraspinal neural stem cell transplantation has been shown to have improved ALS Functional Rating Scale-Revised scores, a standard assessment for ALS (3,4). Clinical trials for ribonucleic acid–targeted therapeutics for genetic variants of ALS also have started (5). Although there is no blood test for ALS, the diagnosis of the disease has evolved. ALS is diagnosed largely on the basis of signs and symptoms as well as neurophysiologic tests, primarily electromyograms.
ALS affects persons of all races and ethnicities. Several potential risk factors for ALS have been identified. Whites, males, non-Hispanics, those aged >60 years, and those with a family history of the disease are more likely to develop ALS (1,6). Previous exposure to heavy metals (e.g., lead and chromium), pesticides, and β-N-methylamino-L-alanine (BMAA) produced by cyanobacteria also have been associated with an increased risk for ALS (7–11). Military service continues to be a possible risk factor for ALS; however, no strong evidence has been found linking etiology with service (12–15). Other possible risk factors such as nutritional intake, use of statins, exposure to viral agents, vigorous physical activity, and trauma also have been identified as possible risk factors (12,16–21).
Background
In 2008, the U.S. Congress passed the ALS Registry Act, which authorized the creation and maintenance of the National ALS Registry (22). The main goals of the Registry are to better describe the incidence and prevalence of ALS, examine risk factors such as environmental and occupational factors, and characterize the demographics of persons living with ALS. As an environmental public health agency affiliated with CDC, the Agency for Toxic Substances and Disease Registry (ATSDR) was designated to establish and maintain the Registry because of ATSDR’s previous pilot work on ALS, the potential environmental risk factor(s) linked to the disease, and its experience with designing other health registries (e.g., the World Trade Center Health Registry, Tremolite Asbestos Registry). ATSDR’s National ALS Registry contributes critical data for further analysis of incidence, prevalence, and the identification of possible risk factors. The Registry employs an innovative use of administrative and self-reported data to identify cases, whereas traditional noncommunicable disease registries typically rely on data from health care providers to identify cases. The novel method underlying the Registry could serve as a model for estimating the prevalence of other nonnotifiable diseases such as Parkinson’s or Alzheimer’s (23).
This report updates the initial Registry findings regarding ALS prevalence for the period October 19, 2010–December 31, 2011 (6) and presents new findings for 2012–2013. The intended audience for this report comprises public health officials, clinicians, and researchers working to better understand ALS etiology and address the needs of persons with ALS and their families.
Methods
The National ALS Registry, established in 2009 and administered by ATDSR, uses a two-pronged approach to identify prevalent cases of ALS in the United States. State- and metropolitan-level surveillance projects also were undertaken to identify incident cases and to evaluate the completeness of the Registry. All incident and prevalent cases are identified for surveillance purposes and not for diagnosis or treatment. This report presents Registry findings related to ALS prevalence only.
The first approach used to identify prevalent cases relies on existing administrative data (from the Centers for Medicare and Medicaid Services, the Veterans Heath Administration [VHA], and the Veterans Benefits Administration [VBA]). A pilot tested algorithm is applied to the administrative data that identifies persons with ALS on the basis of encounter* codes such as having ALS listed as a code in the visit record or having such a code and having seen a neurologist, a death certificate listing ALS as a cause or contributing cause of death, and prescription for Riluzole (Box 1) (24). The second approach, which was launched to the public on October 19, 2010, uses a secure web portal (https://www.cdc.gov/als) to identify cases that are not included in the national administrative databases. This approach allows patients to self-identify and enroll in the National ALS Registry if screening criteria are met. An additional advantage of this approach is those who self-enroll in the Registry can take brief surveys that are used to evaluate possible risk factors for ALS (6). Information is merged into a single record for each person. Merging records for persons identified as having ALS from the administrative databases with those persons who enrolled in the National ALS Registry web portal creates a unique record after data are de-duplicated by using a combination of the last five digits of the person’s Social Security number, sex, month and year of birth, and first and last name. This process ensures that persons who are identified in both the administrative databases and the web portal, and those who have records in multiple years, are not counted twice.
Misclassification of age at diagnosis is likely in the administrative data because the date of diagnosis is not included and must be estimated on the basis of the first date of a medical service encounter. Because of the individual variation in the time lag between diagnosis and benefit approval, using narrow age categories increases the likelihood of miscategorization. For this reason, standard 10-year age categories were used to reduce the potential for misclassification of age at diagnosis.
This report is restricted to prevalence estimates for 2012–2013 and revised prevalence estimates for October 19, 2010–December 31, 2011 that have been updated on the basis of an updated algorithm and death data from the NDI. All activities involving human subjects were reviewed and approved by CDC’s Institutional Review Board (IRB).
Updated Algorithm Used to Identify Cases
The National ALS Registry used an existing algorithm to identify persons with ALS (Figure 1) (Box 1). The algorithm used was developed initially during the pilot projects and categorized cases as “definite ALS,” “possible ALS,” or “not ALS,” with a sensitivity of 87% and a specificity of 85% (24). On the basis of knowledge of ALS and findings from other studies (25–27), individual and combined variables were entered into the algorithm. The variables included International Classification of Diseases Ninth Revision (ICD-9) 335.20 and VBA 8017 codes, a prescription for Riluzole, and the frequency of visits to a neurologist. Only data for persons categorized as “definite ALS” are included in this report. Those determined to be “possible ALS” were not included and will be reevaluated as additional years of data become available.
Persons who chose a Medicare Advantage Plan (similar to a health maintenance organization) do not have individual encounter data and can be missed by the algorithm. Therefore, the first criterion (Box 1) was modified for cases counted in 2012 and 2013 and applied retrospectively to the October 2010–December 2011 dataset. Originally, the criterion required an encounter coded for ALS (ICD-9 code 335.20) in ≥1 years and either a prescription for Riluzole or a death certificate listing ALS as a cause of death. This criterion was modified so that a person who had any two of the three elements (i.e., a prescription for Riluzole and a death certificate listing ALS as a cause of death or a death certificate and an encounter coded for ALS or a prescription for Riluzole and an encounter coded for ALS) was considered to have a definite case of ALS. This modification permits identification of some persons who chose a Medicare Advantage Plan.
Data for those persons ever identified by the algorithm as being “definite” or “possible” ALS patients are submitted to the NDI annually to determine vital status, and if deceased, cause of death. The cause of death is used as part of the algorithm (Box 1). The date of death is used to ensure that the patient is counted toward prevalence only in the appropriate year(s). The updated prevalence estimate for 2010–2011 includes death data that were not available at the time of the first publication (6).
Self-Identification Through a Secure Web Portal
Not all persons with ALS can be identified through the existing national databases because of eligibility requirements for each of the sources as well as potential obstacles to applying for benefits (e.g., economic or educational disadvantage). Therefore, a secure web portal was created to help identify ALS cases not found in the administrative databases. To enter the National ALS Registry through its web portal, patients must answer a series of validation (screening) questions. These validation questions were obtained from the Veterans Administration’s ALS Registry (which is no longer enrolling persons with ALS) and were found to be effective; 93.4% of those who passed the screening questions were determined by a neurologist to have ALS/motor neuron disease (28). Those persons who do not “pass” the ATSDR screening questions cannot register and are instructed to call Registry staff for more information.
Prevalence Calculation
The prevalence of ALS was calculated from the Registry by using the de-duplicated total number of persons with ALS identified through administrative data and those who self-identified for the numerator. The 2011 Census was used for the denominator of the updated 2011 prevalence estimate (29). The 2012 Census estimate was used for the denominator for 2012 (30) and the 2013 Census estimate was used for 2013 (31).
Results
Revised Prevalence, 2010–2011
Due to revisions to the algorithm and the availability of cause-of-death data, an updated prevalence estimate has been calculated for October 19, 2010–December 31, 2011. This updated estimate showed a prevalence rate of 4.3 per 100,000 population and a total of 13,282 cases. This was a slight increase from the initial prevalence estimate of 3.9 per 100,000 persons and 12,187 cases of “definite ALS” (6). Prevalence rates for specific age groups, and for males and females as well as whites and blacks, did not change substantially. The increase in the number of cases was from the national databases, as no additional cases were ascertained from the web portal alone.
Prevalence, 2012
A total of 14,713 persons were identified as “definite ALS” across the four national databases and through web portal registration during 2012 (Table 1). The estimated prevalence rate for 2012 was 4.7 per 100,000 population. The age group 18–39 years had the lowest prevalence rate (0.6 per 100,000 population), and the age group 70–79 had the highest prevalence rate (20.5) (Figure 2). Males continue to have a higher overall prevalence rate (5.9 per 100,000 population) than females (3.5) across each data source individually and overall. The ratio of males to females was 1.6:1. (Figure 3). The prevalence rate for whites was twice that of blacks; whites had a prevalence rate of 5.0 compared with 2.4 for blacks (Figure 4). Information on race was known for 13,336 (90%) of the patients. When all the data sources were combined, whites accounted for the majority of cases (78.6%) followed by unknown race, blacks, and other race (Table 1). Approximately 80% of those identified had administrative data; the proportion of whites who registered was higher in the web portal when compared with the national databases and therefore cases ascertained from the web portal alone might not be representative of all persons with ALS (data not shown).
Prevalence, 2013
A total of 15,908 persons were identified as “definite ALS” across the four national databases and through web portal registration during 2013 (Table 2). The estimated prevalence rate for 2013 was 5.0 per 100,000 population. Persons in the age group 18–39 years had the lowest prevalence rate (0.6 per 100,000 population), and the age group 70–79 years had the highest prevalence rate (20.2) (Figure 2). Males continue to have a higher overall prevalence rate (6.4) than females (3.7); across each data source individually and overall. The ratio of males to females was 1.7:1 (Figure 3). The prevalence rate for whites was twice that of blacks; whites had a prevalence rate of 5.3 per 100,000 population compared with 2.4 for blacks (Figure 4). Information on race was known for 14,124 (89%) of the patients. When all the data sources were combined, whites accounted for the majority of cases (77.4%) followed by unknown race, blacks, and other race. Approximately 80% of those identified had administrative data; the proportion of whites who registered was higher in the web portal when compared with the national databases and therefore cases ascertained from the web portal alone might not be representative of all persons with ALS (data not shown).
Discussion
This report presents updated ALS prevalence estimates for the United States for 2010–2012. Data sources for the Registry remain unchanged: national administrative databases and the online web portal. The Registry’s novel approach of using national administrative databases is the cornerstone in identifying ALS cases because most of the “definite ALS” cases from 2010–2013 originate from these databases. Since publication of the initial surveillance summary report on ALS prevalence (6), the algorithm used to determine “definite ALS” cases has been updated and enhanced, resulting in more cases detected. This algorithm improvement has resulted in an updated prevalence rate from October 19, 2010–December 31, 2011 of 4.3 per 100,000 population and a total of 13,282 cases. This slight increase in ALS prevalence from 3.9 and 12,187 cases as published in the previous report (6) likely reflects better case ascertainment as a result of the revised algorithm. For calendar years 2012 and 2013, prevalence rates were 4.7 and 5.0, respectively. A total of 14,713 cases of “definite ALS” was reported for 2012 and 15,908 for 2013. The reason for the increase in ALS cases might be attributed to the revised algorithm to determine “definite” ALS cases as well as increased public awareness. For all surveillance periods (2010–2013), ALS is more common in whites, males, and those aged ≥60 years. The pattern of characteristics (e.g., age and sex) for persons with ALS identified in 2010–2011 was similar to persons identified in 2012 and 2013.
Increased awareness of the National ALS Registry through improved public education and outreach by ATSDR and partner organizations also could have resulted in more persons registering in the web portal, thereby resulting in an increased prevalence estimate. Use of social media channels such as Facebook and Twitter have raised awareness and is indicative of increased web portal enrollment for 2012 and 2013, and during 2010–2011 (i.e., more patients continue to visit the Registry and convert to registrants). For example, during the summer 2014 ALS Ice-Bucket Challenge, Registry enrollment and web traffic increased from the same period the year before by 8% and 45%, respectively. Social media phenomena such as the ALS Ice Bucket Challenge highlight and promote awareness and increased attention for this disease.
If the number of ALS cases continues to increase over time, another explanation might be the increased use of multidisciplinary ALS centers across the United States, leading to longer survivorship (27,32–34). These centers centralize ALS multidisciplinary care for patients bringing together neurologists; occupational, speech, and physical therapists; nutritionists; pharmacists; and social workers to provide specialized care in one place and at one time (35). As ALS patients live longer, ALS prevalence might possibly increase slightly. Additional data are needed to validate this hypothesis.
ALS affects whites and males at a higher rate and remains unchanged across all data sources and years. The age groups 60–69 years and 70–79 years are the most common ages of diagnosis and those aged 18–39 years are the least. This finding is consistent with reported literature in the United States and abroad (1,6,36–43).
Because ALS is not a nationally notifiable disease (i.e., it is not a condition in which federal agencies are notified by local and state health jurisdictions upon diagnosis), the Registry is attempting to estimate the number of missing ALS cases in the United States. A statistical approach to determine the completeness of the Registry is capture-recapture analysis, which is a method for estimating the maximum population with a disease based on the “capture” of specific persons’ different sources (44–46). This technique has been utilized previously on a smaller and national geographic scale for the under-reporting of ALS cases (47,48). Using capture-recapture on a national level for the administrative databases and portal data is currently underway by ATSDR to estimate the completeness of the Registry.
Additional data are needed to determine trends for ALS prevalence in the United States. Once data from calendar years 2014 and 2015 are available, it will be possible to monitor and characterize the variability of prevalence over time. Currently, there is no indication that the prevalence of ALS is increasing in the United States. ATSDR is currently analyzing ALS death data from 2011–2013 in order to estimate the ALS mortality rate in the United States.
To determine localized incidence and prevalence of ALS, evaluate the completeness of the Registry, and better describe the demographics of persons with ALS, ATSDR funded state and metropolitan (metro) level surveillance projects in three states (Florida, New Jersey, and Texas) and in eight geographic metro areas (Atlanta, Georgia; Baltimore, Maryland; Chicago, Illinois; Detroit, Michigan; Las Vegas, Nevada; Los Angeles, California; Philadelphia, Pennsylvania; and San Francisco, California) covering 2009–2011. Because ALS is predominantly a disease that affects whites, these states and metro areas were selected for their over-representation of minority populations of blacks, Asians, and Hispanics. Incidence rates ranged from 1.1 to 2.1 per 100,000 person-years in individual states and metro areas (39,41,49–51) and the incidence rate was 1.5 person-years for all states and metro areas combined (52). Incidence rates by race and ethnicity were calculated for all states and metro areas combined and showed that the incidence rate in whites was higher than the incidence rates for blacks and Asians. In addition, the incidence rate in non-Hispanics was significantly higher than the incidence rate in Hispanics (40). Because the states and metro areas were selected to over-represent racial and ethnic minorities, the differences in incidence rates between the individual states and metro areas could be related to the differences in the racial and ethnic make-up of the underlining populations. The overall prevalence rate for the states and metro areas combined was 3.8 per 100,000 population (52), which is consistent with current literature (42,43) and comparable to the original Registry prevalence estimate of 3.9 per 100,000 (6). Data from the state and metro area-based surveillance are being compared with data from the same areas collected by the Registry to determine if any geographic areas or subgroups are underrepresented.
ATSDR is committed to helping the scientific community better understand the etiology of ALS. Since October 2012, ATSDR has funded 13 external research studies, of which five were investigator-initiated R01 studies funded during 2015–2016. Research topics ranged from case-control studies, examining environmental risk factors for ALS, whether certain medical conditions or drugs might affect ALS susceptibility, as well as others. A complete list of funded research is available at https://wwwn.cdc.gov/als/ALSExternalResearchfundedbyRegistry.aspx).
In addition to monitoring the prevalence of ALS in the United States, the National ALS Registry also is examining risk factors for the disease. Currently 17 risk factor modules are available online through the web portal that patients can complete (Box 2). Partial results from the first six surveys have been published (6,53), and analyses for other surveys are underway. To date, approximately 50,000 survey modules have been completed by Registry enrollees.
The quantification of a nonnotifiable disease such as ALS continues to be a challenge. The National ALS Registry model of using national administrative databases and an online portal is a novel method for estimating prevalence and could be used for other nonnotifiable diseases (23).
Limitations
The findings in this report are subject to at least four limitations. First, because ALS is not a notifiable disease, ensuring that all newly diagnosed and prevalent ALS cases in the United States are captured in the Registry is challenging and therefore the possibility of under-ascertainment exists. However, the large administrative database methodology that ATSDR is using was vetted through a pilot effort and is expected to identify most of the ALS cases in the United States, given its high sensitivity and specificity (24). In addition, ATSDR is partnering with national stakeholders to promote the Registry to persons with the disease so they can self-enroll though the Registry’s web portal. Second, although every attempt was made to de-duplicate the files, differences in fields collected by the different sources, misspellings of names, and data entry errors could have prevented records from merging correctly. However, it is unlikely that this occurred in numbers sufficient to affect the overall conclusions. Third, the calculation of ALS incidence with Registry data is not possible at this time because the date of diagnosis is not captured through the large administrative database approach, and cases without a date of diagnosis comprise 68% of cases in the Registry. However, through a separate Registry project, incidence has been calculated and the findings published for ALS incidence in smaller defined geographic areas of the United States (39,41,49–52). Finally, the Registry has been officially active since October 2009 and is still being enhanced. As more persons with ALS enroll and complete surveys, a better understanding of possible risk factors might emerge.
Ongoing Registry Enhancements
In addition to addressing the main goals of the Registry outlined by the ALS Registry Act (i.e., describing the incidence and prevalence of ALS, characterizing the demographics of those living with ALS, and examining risk factors), ATSDR also is taking steps to further enhance the Registry for patients and researchers. For example, in 2015, the Registry concluded the National ALS Biorepository Pilot Study. An external group of experts comprising neurologists, researchers, laboratorians, and biorepository experts met and discussed the findings from the pilot study. This group deemed it feasible to launch a nationally representative biorepository as part of the National ALS Registry and indicated that such a biorepository is warranted. It is anticipated that the Biorepository will be launched officially in the fall of 2016. The newly established Biorepository will be novel in several ways. First, it will store samples from in-home collection (e.g., blood, hair, or saliva) and postmortem collection (e.g., brain, bone, spinal cord, cerebrospinal fluid, muscle, and skin). Currently, the few existing ALS biorepositories largely have samples that are from specific clinics or medical practices and/or the samples are left over from previous clinical trials in the United States. Specimens from the National ALS Biorepository will be collected from a geographically representative sample of persons with ALS who are enrolled in the Registry. The sample of persons recruited to participate will correlate to the population distribution of the United States and each year will include at least one person from each state. The exact number of ALS registrants with specimens collected will vary from year to year depending on the number of persons to be recruited. These de-identified samples can then be paired with completed risk factor survey data. Researchers will be able to request a sample as-is or paired with risk factor data. Premortem biospecimens will be collected in a participant’s home by a trained phlebotomist. The Biorepository will not charge participants for providing specimens. The availability of additional specimens on a national sample of ALS patients will further expand research on the genetics, potential biomarkers, and etiology for ALS. Researchers can request samples from the National ALS Biorepository for studies that have IRB approval. Once submitted, ATSDR will review the application for completeness, sample availability, and relevance to ALS research. The application will subsequently be evaluated by an internal/external scientific panel. ATSDR estimates that the approval process could take up to 60 business days from the receipt of a complete application. More information about biological samples and tissues is available by calling 1-855-874-6912 (Monday through Friday 8:30 am to 5:00 pm ET).
ATSDR also is preparing to incorporate the National Institutes of Health’s Global Unique Identifier (GUID) system into the Registry. The GUID will allow the generation of a unique alphanumeric code which is specific to the participant and will allow de-identified subjects to be tracked over time across various clinical trials, research studies, databases, and even biobanks (54). During the Registry enrollment process, registrants will be able to consent and provide the necessary information to participate in the GUID system voluntarily.
The Registry’s research notification mechanism informs enrolled persons with ALS about new clinical trials and research studies in which they might be eligible. When researchers send ATSDR information about their studies, an internal/external review panel evaluates the project and verifies that the study has been approved by the researcher’s IRB. ATSDR then sends information about the study via e-mail to Registry enrollees who have agreed to be contacted about such projects. Registrants then can contact the researcher if they want to take part in the study. ATSDR does not provide identifiable information to researchers at any point in this process. Since the research notification mechanism’s May 2012 deployment, approximately 96% of enrollees in the National ALS Registry have elected to be notified about ALS research opportunities. To date, approximately 60,000 email notifications have been sent to Registry enrollees for 23 clinical trials and epidemiological studies (https://wwwn.cdc.gov/als/ALSResearchNotificationClinicalTrialsStudies.aspx).
Conclusion
Public health surveillance is important to determine the epidemiology of rare diseases such as ALS. Surveillance data from the National ALS Registry continue to be used for determining the national, state, and metropolitan estimates of incidence and prevalence, as well as assessing the potential risk factor(s) and etiology of ALS. In addition, the Registry is a source to voluntarily match enrollees with research studies and clinical trials.
This is the second report on the prevalence estimate of ALS for the United States based on national data and covers two individual calendar years. The establishment of the National ALS Registry, as well as the upcoming launch of the National ALS Biorepository, fills a critical scientific gap by providing prevalence estimates of this disease and facilitates further study of risk factors and etiology. Using existing database resources from the Centers for Medicare and Medicaid Services, the Veterans Heath Administration, and the Veterans Benefits Administration, as well as the self-reported web-based portal is allowing for more accurate prevalence estimates and could be a model for other non-notifiable diseases such as Parkinson’s disease or multiple sclerosis (23). The National ALS Registry continues to be improved and have enhancements added that will expand ALS research as well as identify and detect more ALS cases. ATSDR is committed to advancing ALS research and monitoring trends of ALS prevalence in the United States.
Corresponding author: Paul Mehta, MD, Division of Toxicology and Human Health Sciences, Agency for Toxic Substances and Disease Registry. Telephone: 770-488-0556; E-mail: pum4@cdc.gov.
1Agency for Toxic Substances and Disease Registry; 2McKing Consulting Corporation, Atlanta, Georgia; 3Carter Consulting, Inc., Atlanta, Georgia
References
- Mitsumoto HCD, Pioro EP. Amyotrophic lateral sclerosis. Philadelphia, PA: F.A. Davis Company; 1998.
- Carlesi C, Pasquali L, Piazza S, et al. Strategies for clinical approach to neurodegeneration in amyotrophic lateral sclerosis. Arch Ital Biol 2011;149:151–67. PubMed
- Feldman EL, Boulis NM, Hur J, et al. Intraspinal neural stem cell transplantation in amyotrophic lateral sclerosis: phase 1 trial outcomes. Ann Neurol 2014;75:363–73. CrossRef PubMed
- Glass JD, Boulis NM, Johe K, et al. Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells 2012;30:1144–51. CrossRef PubMed
- Riboldi G, Zanetta C, Ranieri M, et al. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol Neurobiol 2014;50:721–32. CrossRef PubMed
- Mehta P, Antao V, Kaye W, et al. Prevalence of amyotrophic lateral sclerosis—United States, 2010–2011. MMWR Suppl 2014;63(No. SS-7). PubMed
- Cavaleri F. Review of amyotrophic lateral sclerosis, Parkinson’s and Alzheimer’s diseases helps further define pathology of the novel paradigm for Alzheimer’s with heavy metals as primary disease cause. Med Hypotheses 2015;85:779–90. CrossRef PubMed
- Wang MD, Gomes J, Cashman NR, Little J, Krewski D. A meta-analysis of observational studies of the association between chronic occupational exposure to lead and amyotrophic lateral sclerosis. J Occup Environ Med 2014;56:1235–42. CrossRef PubMed
- Bradley WG, Borenstein AR, Nelson LM, et al. Is exposure to cyanobacteria an environmental risk factor for amyotrophic lateral sclerosis and other neurodegenerative diseases? Amyotroph Lateral Scler Frontotemporal Degener 2013;14:325–33. CrossRef PubMed
- Roelofs-Iverson RA, Mulder DW, Elveback LR, Kurland LT, Molgaard CA. ALS and heavy metals: a pilot case-control study. Neurology 1984;34:393–5. CrossRef PubMed
- Banack SA, Metcalf JS, Bradley WG, Cox PA. Detection of cyanobacterial neurotoxin β-N-methylamino-l-alanine within shellfish in the diet of an ALS patient in Florida. Toxicon 2014;90:167–73. CrossRef PubMed
- Oskarsson B, Horton DK, Mitsumoto H. Potential environmental factors in amyotrophic lateral sclerosis. Neurol Clin 2015;33:877–88. CrossRef PubMed
- Weisskopf MG, Cudkowicz ME, Johnson N. Military service and amyotrophic lateral sclerosis in a population-based cohort. Epidemiology 2015;26:831–8. CrossRef PubMed
- Beard JD, Kamel F. Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis etiology and survival. Epidemiol Rev 2015;37:55–70. CrossRef PubMed
- Capozzella A, Sacco C, Chighine A, et al. Work related etiology of amyotrophic lateral sclerosis (ALS): a meta-analysis. Ann Ig 2014;26:456–72. PubMed
- Okamoto K, Kihira T, Kobashi G, et al. Fruit and vegetable intake and risk of amyotrophic lateral sclerosis in Japan. Neuroepidemiology 2009;32:251–6. CrossRef PubMed
- Factor-Litvak P, Al-Chalabi A, Ascherio A, et al. Current pathways for epidemiological research in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2013;14(Suppl 1):33–43. CrossRef PubMed
- Li W, Lee MH, Henderson L, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 2015;7:307ra153. CrossRef PubMed
- Fang F, Chen H, Wirdefeldt K, et al. Infection of the central nervous system, sepsis and amyotrophic lateral sclerosis. PLoS One 2011;6:e29749. CrossRef PubMed
- Eaglehouse YL, Talbott EO, Chang Y, Kuller LH. Participation in physical activity and risk for amyotrophic lateral sclerosis mortality among postmenopausal women. JAMA Neurol 2016;73:329–36. CrossRef PubMed
- Beghi E, Logroscino G, Chiò A, et al. Amyotrophic lateral sclerosis, physical exercise, trauma and sports: results of a population-based pilot case-control study. Amyotroph Lateral Scler 2010;11:289–92. CrossRef PubMed
- US Public Health Service. ALS Registry Act. Washington, DC: 110th Congress. Public Law 2008;122 Stat 4047:110–373.
- Horton DK, Mehta P, Antao VC. Quantifying a nonnotifiable disease in the United States: the National Amyotrophic Lateral Sclerosis Registry model. JAMA 2014;312:1097–8. CrossRef PubMed
- Kaye WE, Sanchez M, Wu J. Feasibility of creating a national ALS registry using administrative data in the United States. Amyotroph Lateral Scler Frontotemporal Degener 2014;15:433–9. CrossRef PubMed
- Fisher ES, Baron JA, Malenka DJ, Barrett J, Bubolz TA. Overcoming potential pitfalls in the use of Medicare data for epidemiologic research. Am J Public Health 1990;80:1487–90. CrossRef PubMed
- Pope GC, Urato CJ, Kulas ED, Kronick R, Gilmer T. Prevalence, expenditures, utilization, and payment for persons with MS in insured populations. Neurology 2002;58:37–43. CrossRef PubMed
- Taylor DH , Fillenbaum GG, Ezell ME. The accuracy of Medicare claims data in identifying Alzheimer’s disease. J Clin Epidemiol 2002;55:929–37. CrossRef PubMed
- Allen KD, Kasarskis EJ, Bedlack RS, et al. The National Registry of Veterans with Amyotrophic Lateral Sclerosis. Neuroepidemiology 2008;30:180–90. CrossRef PubMed
- US Census Bureau. American Community Survey 1-year estimates. B21001–sex by age by veterans status for civilian population 18 years plus. Washington, DC: US Census Bureau; 2011.
- US Census Bureau. Total population 2012 American Community Survey 1-year. Washington, DC: US Census Bureau; 2012.
- US Census Bureau. Annual estimates of the resident population for the United States, regions, states, and Puerto Rico: April 1, 2010 to July 1, 2013 (NST-EST2013–01). Washington, DC: US Census Bureau; 2013.
- Chiò A, Bottacchi E, Buffa C, Mutani R, Mora G; PARALS. Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. J Neurol Neurosurg Psychiatry 2006;77:948–50. CrossRef PubMed
- Van den Berg JP, Kalmijn S, Lindeman E, et al. Multidisciplinary ALS care improves quality of life in patients with ALS. Neurology 2005;65:1264–7. CrossRef PubMed
- Boylan K, Levine T, Lomen-Hoerth C, et al. ; ALS Center Cost Evaluation W/Standards & Satisfaction (Access) Consortium. Prospective study of cost of care at multidisciplinary ALS centers adhering to American Academy of Neurology (AAN) ALS practice parameters. Amyotroph Lateral Scler Frontotemporal Degener 2015;17:119–27. CrossRef PubMed
- Amyotrophic Lateral Sclerosis Association. ALS multidisciplinary clinics. Chicago, IL: Amyotrophic Lateral Sclerosis Association, Greater Chicago Chapter. http://webchicago.alsa.org/site/PageNavigator/CHI_8_Clinics.html
- Couratier P, Corcia P, Lautrette G, Nicol M, Preux PM, Marin B. Epidemiology of amyotrophic lateral sclerosis: a review of literature. Rev Neurol (Paris) 2016;172:37–45. CrossRef PubMed
- Wagner L, Archer NP, Williamson DM, Henry JP, Schiffer R, Jackson CE. Prevalence of amyotrophic lateral sclerosis in Texas, 1998–2003. Tex Med 2012;108:e1. PubMed
- Turabelidze G, Zhu BP, Schootman M, et al. An epidemiologic investigation of amyotrophic lateral sclerosis in Jefferson County, Missouri, 1998–2002. Neurotoxicology 2008;29:81–6. CrossRef PubMed
- Jordan H, Fagliano J, Rechtman L, Lefkowitz D, Kaye W. Population-based surveillance of amyotrophic lateral sclerosis in New Jersey, 2009–2011. Neuroepidemiology 2014;43:49–56. CrossRef PubMed
- Rechtman L, Jordan H, Wagner L, Horton DK, Kaye W. Racial and ethnic differences among amyotrophic lateral sclerosis cases in the United States. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:65–71. CrossRef PubMed
- Jordan H, Rechtman L, Wagner L, Kaye WE. Amyotrophic lateral sclerosis surveillance in Baltimore and Philadelphia. Muscle Nerve 2015;51:815–21. CrossRef PubMed
- Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. How common are the ‘common’ neurologic disorders? Neurology 2007;68:326–37.
- Chio A, Logrosino G, Traynor BJ, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013;41:118–30.
- Tilling K. Capture-recapture methods—useful or misleading? Int J Epidemiol 2001;30:12–4. CrossRef PubMed
- Robles SC, Marrett LD, Clarke EA, Risch HA. An application of capture-recapture methods to the estimation of completeness of cancer registration. J Clin Epidemiol 1988;41:495–501. CrossRef PubMed
- Schouten LJ, Straatman H, Kiemeney LA, Gimbrère CH, Verbeek AL. The capture-recapture method for estimation of cancer registry completeness: a useful tool? Int J Epidemiol 1994;23:1111–6. CrossRef PubMed
- Wittie M, Nelson LM, Usher S, Ward K, Benatar M. Utility of capture-recapture methodology to assess completeness of amyotrophic lateral sclerosis case ascertainment. Neuroepidemiology 2013;40:133–41. CrossRef PubMed
- Huisman MH, de Jong SW, van Doormaal PT, et al. Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J Neurol Neurosurg Psychiatry 2011;82:1165–70. CrossRef PubMed
- Rechtman L, Wagner L, Kaye W, Villanacci J. Updated Prevalence and Demographic Characteristics for ALS Cases in Texas, 2009–2011. South Med J 2015;108:483–6. PubMed
- Freer C, Hylton T, Jordan HM, Kaye WE, Singh S, Huang Y. Results of Florida’s Amyotrophic Lateral Sclerosis Surveillance Project, 2009–2011. BMJ Open 2015;5:e007359. CrossRef PubMed
- Valle J, Roberts E, Paulukonis S, Collins N, English P, Kaye W. Epidemiology and surveillance of amyotrophic lateral sclerosis in two large metropolitan areas in California. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:209–15. CrossRef PubMed
- Wagner L, Rechtman L, Jordan H, et al. State and metropolitan area-based amyotrophic lateral sclerosis (ALS) surveillance. Amyotroph Lateral Scler Frontotemporal Degener 2015;17:128–34. CrossRef PubMed
- Bryan L, Kaye W, Antao V, Mehta P, Muravov O, Horton DK. Preliminary results of National Amyotrophic Lateral Sclerosis (ALS) Registry risk factor survey data. PLoS One 2016;11:e0153683. CrossRef PubMed
- National Institutes of Health. Global Unique Identifier (GUID). Bethesda, MD: National Institutes of Health; 2016. https://ncats.nih.gov/grdr/guid
* An encounter is a record of a health care service (e.g., a visit to a physician, hospitalization, x-ray, or laboratory test.
 BOX 1. Algorithm used to identify cases of amyotrophic lateral sclerosis
BOX 1. Algorithm used to identify cases of amyotrophic lateral sclerosis
The Agency for Toxic Substances and Disease Registry applied an updated algorithm developed as part of the four pilot projects to the same data described in this report to identify persons with amyotrophic lateral sclerosis (ALS) during 2012–2013. The algorithm categorizes persons into three categories: “definite ALS,” “possible ALS,” and “not ALS.”
Those included in the “definite ALS” category met one or more of the following criteria:
-
any two of the following: 1) an encounter* coded for ALS (International Classification of Diseases, Ninth Revision [ICD-9]335.20) in 1 or more years in the same source, or 2)a death certificate listing ALS as a cause of death, or 3) a prescription for Riluzole; OR
-
an encounter coded for ALS (ICD-9 335.20) in ≥2 years for which at least one visit must be with a neurologist visit in the same source; OR
-
a person aged <65 years with an encounter coded for ALS (ICD-9 335.20) in Medicare where at least one visit is with a neurologist visit; OR
-
an encounter coded for ALS (ICD-9 335.20) in ≥1 year(s) for which at least one visit must be with a neurologist visit in the same source and an encounter coded for ALS in another source; OR
-
an encounter coded for ALS (ICD-9 335.20 or Veterans Benefits Administration 8017 codes) in three or more sources; OR
-
an encounter coded for ALS (ICD-9 335.20) in 1 year and five or more neurologist visits in the same source.
Those included in the “not ALS” category met one or more of the following criteria:
-
no encounter coded for ALS (ICD-9 335.20) in any source and no prescription for Riluzole; OR
-
an encounter coded for ALS (ICD-9 335.20) in only 1 year but no neurologist visit in the same source; OR
-
a person aged <18 years.
Persons in the “possible ALS” category include everyone not determined to be either “definite ALS” or “not ALS.”
* A record of a health care service (e.g., a visit to a physician, hospitalization, x-ray, or laboratory test).
FIGURE 1. National ALS Registry methodology*
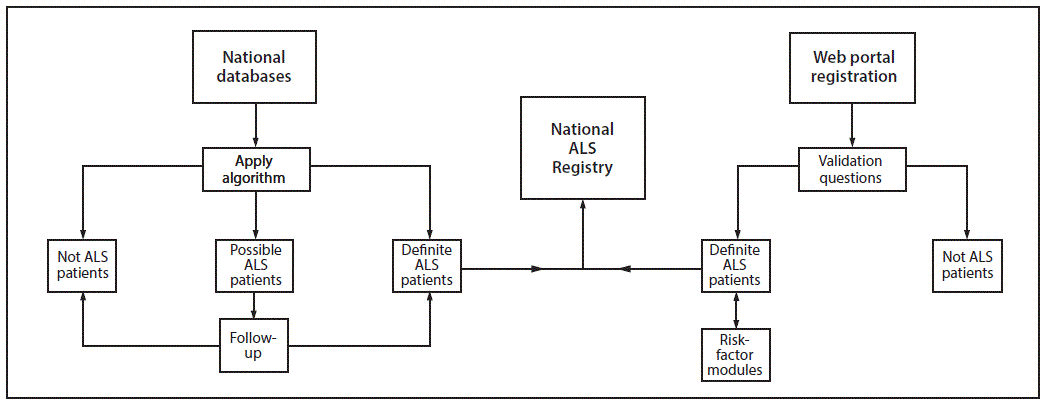
Abbreviation: ALS = amyotrophic lateral sclerosis.
* National databases are maintained by Medicare, Medicaid, the Veterans Health Administration, and the Veterans Benefit Administration. Web portal registration is available at http://wwwn.cdc.gov/als.
TABLE 1. Number and percentage of identified cases of amyotrophic lateral sclerosis, by age group, sex, and race — National ALS Registry, United States, 2012
| Characteristic | Total | Census data* | Prevalence estimate | 95% CI |
|---|---|---|---|---|
| No. (%) | ||||
| Age group (yrs) | ||||
| 18–39 | 527 (3.6) | 93,147,582 | 0.6 | 0.52–0.61 |
| 40–49 | 1,535 (10.4) | 42,801,650 | 3.6 | 3.41–3.77 |
| 50–59 | 3,079 (20.9) | 43,148,991 | 7.1 | 6.88–7.39 |
| 60–69 | 4,431 (30.1) | 31,857,017 | 13.9 | 13.50–14.32 |
| 70–79 | 3,604 (24.5) | 17,603,434 | 20.5 | 19.80–21.14 |
| ≥80 | 1,480 (10.1) | 11,644,956 | 12.7 | 12.06–13.36 |
| Unknown† | 57 (0.4) | NA | NA | NA |
| Sex | ||||
| Male | 9,043 (61.5) | 154,436,243 | 5.9 | 5.73–5.98 |
| Female | 5,649 (38.4) | 159,477,797 | 3.5 | 3.45–3.63 |
| Unknown† | 21 (0.1) | NA | NA | NA |
| Race§ | ||||
| White | 11,565 (78.6) | 231,992,377 | 5.0 | 4.89–5.08 |
| Black | 933 (6.3) | 39,623,138 | 2.4 | 2.20–2.51 |
| Other | 838 (5.7) | NA | NA | NA |
| Unknown | 1,377 (9.4) | NA | NA | NA |
| Total | 14,713 (100.0) | 313,914,040 | 4.7 | 4.61–4.76 |
Abbreviations: ALS = amyotrophic lateral sclerosis; CI = confidence interval; NA = not applicable.
*Sources: B01001: Sex by age. 2012 American Community Survey 1-year estimates (http://factfinder.census.gov/faces/tableservices/jsf/pages/productview.xhtml?pid=ACS_12_1YR_B01001&prodType=table); B02001: Race. 2012 American Community Survey 1-year estimates (http://factfinder.census.gov/faces/tableservices/jsf/pages/productview.xhtml?pid=ACS_12_1YR_B02001&prodType=table).
† Missing age or sex.
§ Race was defined as either identifying with one racial group or, if racial identification included more than one racial group, respondents were classified as “Other.” If only Hispanic ethnicity and no racial group were chosen, or if Race = Don’t Know, race was defined as “Unknown.”
FIGURE 2. Prevalence rates* for cases of amyotrophic lateral sclerosis, by age group — National ALS Registry, United States, 2012 and 2013
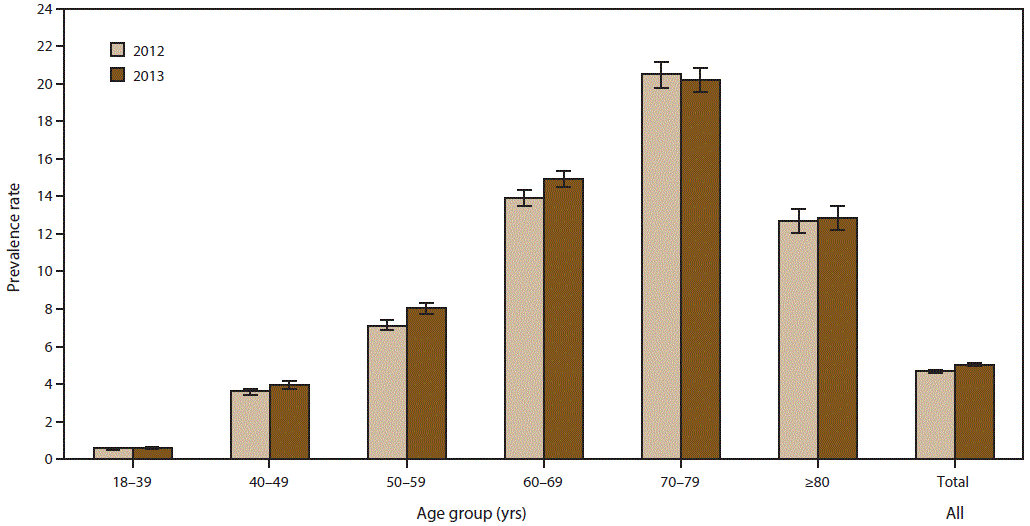
Abbreviation: ALS = amyotrophic lateral sclerosis.
* Per 100,000 population. Bars represent 95% confidence intervals. For 2012, N = 14,713; for 2013, N = 15,908.
FIGURE 3. Prevalence rates* for cases of amyotrophic lateral sclerosis, by sex — National ALS Registry, United States, 2012 and 2013
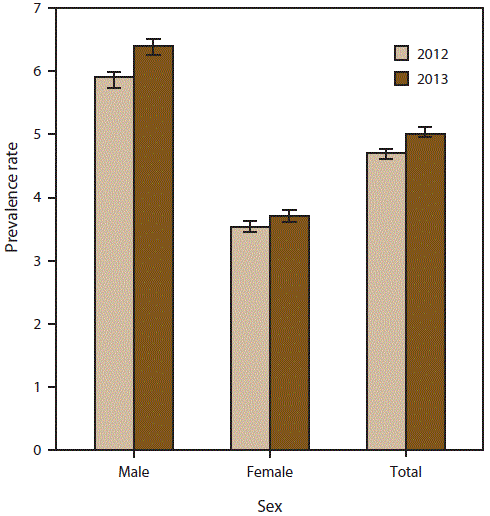
Abbreviation: ALS = amyotrophic lateral sclerosis.
* Per 100,000 population. Bars represent 95% confidence intervals. For 2012, N = 14,713; for 2013, N = 15,908.
FIGURE 4. Prevalence rates* for cases of amyotrophic lateral sclerosis, by race — National ALS Registry, United States, 2012 and 2013
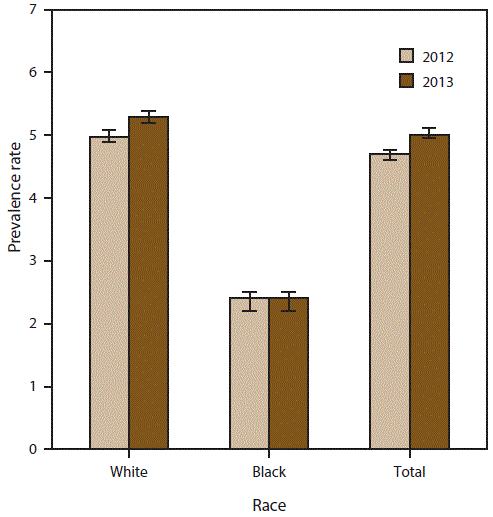
Abbreviation: ALS = amyotrophic lateral sclerosis.
* Per 100,000 population. Bars represent 95% confidence intervals. For 2012, N = 14,713; for 2013, N = 15,908.
TABLE 2. Number and percentage of identified cases of amyotrophic lateral sclerosis, by age group, sex, and race — National ALS Registry, United States, 2013
| Characteristic | Total | Census data | Prevalence estimate | 95% CI |
|---|---|---|---|---|
| No. (%) | ||||
| Age group (yrs) | ||||
| 18–39 | 567 (3.6) | 93,966,985 | 0.6 | 0.55–0.65 |
| 40–49 | 1,667 (10.5) | 42,072,696 | 4.0 | 3.77–4.15 |
| 50–59 | 3,502 (22.0) | 43,609,956 | 8.0 | 7.76–8.30 |
| 60–69 | 4,908 (30.9) | 32,830,297 | 14.9 | 14.53–15.37 |
| 70–79 | 3,695 (23.2) | 18,291,069 | 20.2 | 19.55–20.85 |
| ≥80 | 1,513 (9.5) | 11,771,224 | 12.9 | 12.21–13.50 |
| Unknown† | 56 (0.4) | NA | NA | NA |
| Sex | ||||
| Male | 9,941 (62.5) | 155,627,698 | 6.4 | 6.26–6.51 |
| Female | 5,947 (37.4) | 160,501,141 | 3.7 | 3.61–3.80 |
| Unknown† | 20 (0.1) | NA | NA | NA |
| Race§ | ||||
| White | 12,318 (77.4) | 232,969,901 | 5.3 | 5.19–5.38 |
| Black | 940 (5.9) | 39,919,371 | 2.4 | 2.20–2.51 |
| Other | 866 (5.4) | NA | NA | NA |
| Unknown | 1,784 (11.2) | NA | NA | NA |
| Total | 15,908 (100.0) | 316,128,839 | 5.0 | 4.95–5.11 |
Abbreviations: ALS = amyotrophic lateral sclerosis; CI = confidence interval; NA = not applicable.
*Sources: B01001: Sex by age. 2013 American Community Survey 1-Year Estimates (http://factfinder.census.gov/faces/tableservices/jsf/pages/productview.xhtml?pid=ACS_13_1YR_B01001&prodType=table).; B02001: Race. 2013 American Community Survey 1-Year Estimates (http://factfinder.census.gov/faces/tableservices/jsf/pages/productview.xhtml?pid=ACS_13_1YR_B02001&prodType=table).
† Missing age or sex.
§ Race was defined as either identifying with one racial group or, if racial identification included more than one racial group, respondents were classified as “Other.” If only Hispanic ethnicity and no racial group were chosen, or if Race = Don’t Know, race was defined as “Unknown.”
BOX 2. Current National ALS Registry risk factor surveys
-
Demographics
-
Occupational history
-
Military history
-
Smoking and alcohol history
-
Physical activity
-
Family history of neurologic diseases
-
Disease progression (ALSFRS)
-
Open-ended etiologic questions
-
Clinical data (e.g., date of symptom onset, body part where weakness first noticed)
-
Lifetime residential history
-
Lifetime occupational history
-
Residential pesticide use
-
Hobbies with toxicant exposures
-
Reproductive history (women only)
-
Caffeine consumption
-
Trauma history
-
Health insurance status
Abbreviations: ALS = amyotrophic lateral sclerosis: ALSFRS = ALS Functional Rating Scale.
Suggested citation for this article: Mehta P, Kaye W, Bryan L, et al. Prevalence of Amyotrophic Lateral Sclerosis — United States, 2012–2013. MMWR Surveill Summ 2016;65(No. SS-8):1–12. DOI: http://dx.doi.org/10.15585/mmwr.ss6508a1.
Use of trade names and commercial sources is for identification only and does not imply endorsement by the U.S. Department of
Health and Human Services.
References to non-CDC sites on the Internet are
provided as a service to MMWR readers and do not constitute or imply
endorsement of these organizations or their programs by CDC or the U.S.
Department of Health and Human Services. CDC is not responsible for the content
of pages found at these sites. URL addresses listed in MMWR were current as of
the date of publication.
All HTML versions of MMWR articles are generated from final proofs through an automated process. This conversion might result in character translation or format errors in the HTML version. Users are referred to the electronic PDF version (https://www.cdc.gov/mmwr) and/or the original MMWR paper copy for printable versions of official text, figures, and tables.
Questions or messages regarding errors in formatting should be addressed to mmwrq@cdc.gov.
- Page last reviewed: June 21, 2017
- Page last updated: June 21, 2017
- Content source: